

La mission du Groupe de Modélisation Moléculaire de BioCIS est de soutenir la conception, la découverte et le développement de nouvelles molécules à visée thérapeutique par des approches de biophysique moléculaire computationnelle, incluant les calculs d’amarrage moléculaire, les criblages virtuels, les simulations de dynamique moléculaire tout-atome et gros-grain. Les trois principales thématiques de recherche sont (i) la modélisation des structures de protéines et complexes protéine-ligand, (ii) l’auto-assemblage de biopolymères pour l’encapsulation de principes actifs, et (iii) la conception d’inhibiteurs peptidomimétiques d’interactions protéine-protéine.
Contact : Pr Tâp HA DUONG
Interactions Protéine-Ligand
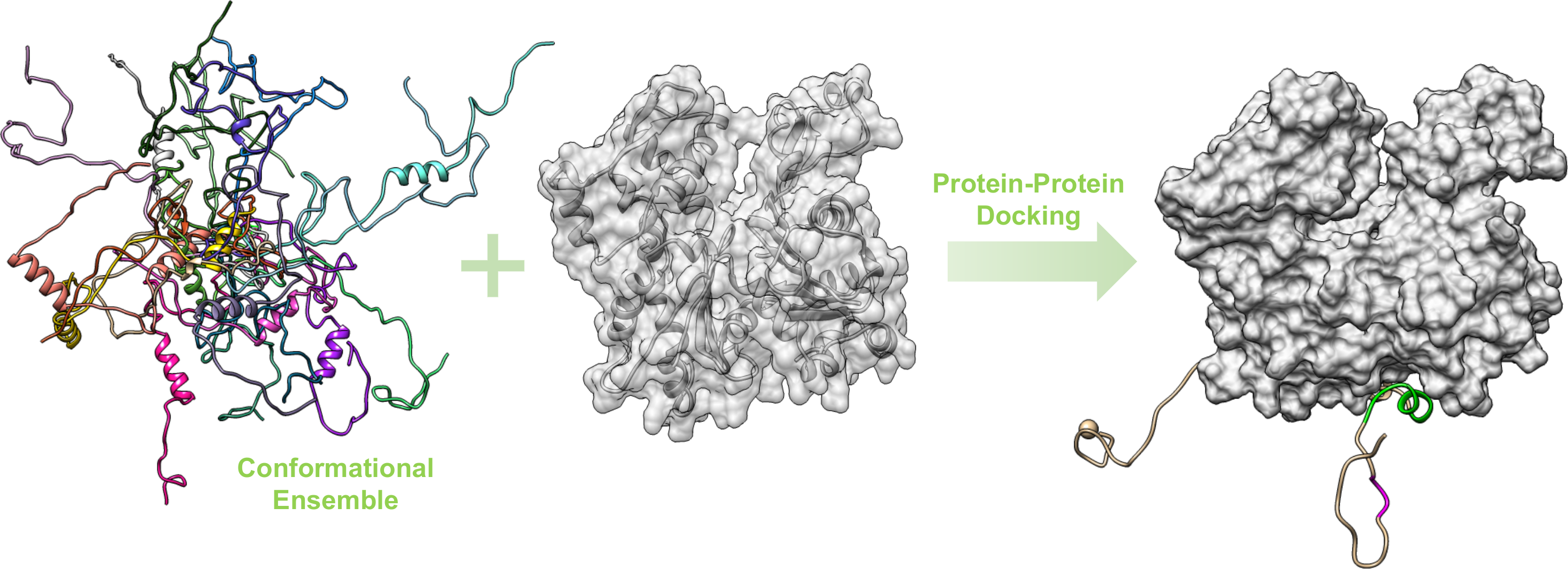
La conception de médicaments basée sur la structure est la principale stratégie utilisée dans notre groupe de modélisation moléculaire pour soutenir la recherche et le développement de nouveaux composés thérapeutiques par les chimistes médicinaux de BioCIS. Ainsi, l’une de nos principales activités consiste à étudier les structures tridimensionnelles des cibles des médicaments, qui sont principalement des protéines et des interactions protéine-protéine (IPP). Nous sommes particulièrement intéressés par les protéines intrinsèquement désordonnées et leurs complexes dont les ensembles conformationnels sont souvent difficiles à caractériser expérimentalement. À cette fin, nous utilisons généralement des simulations de dynamique moléculaire classiques ou améliorées, ainsi que des calculs de docking protéine-protéine.

Auto-assemblage de biopolymères
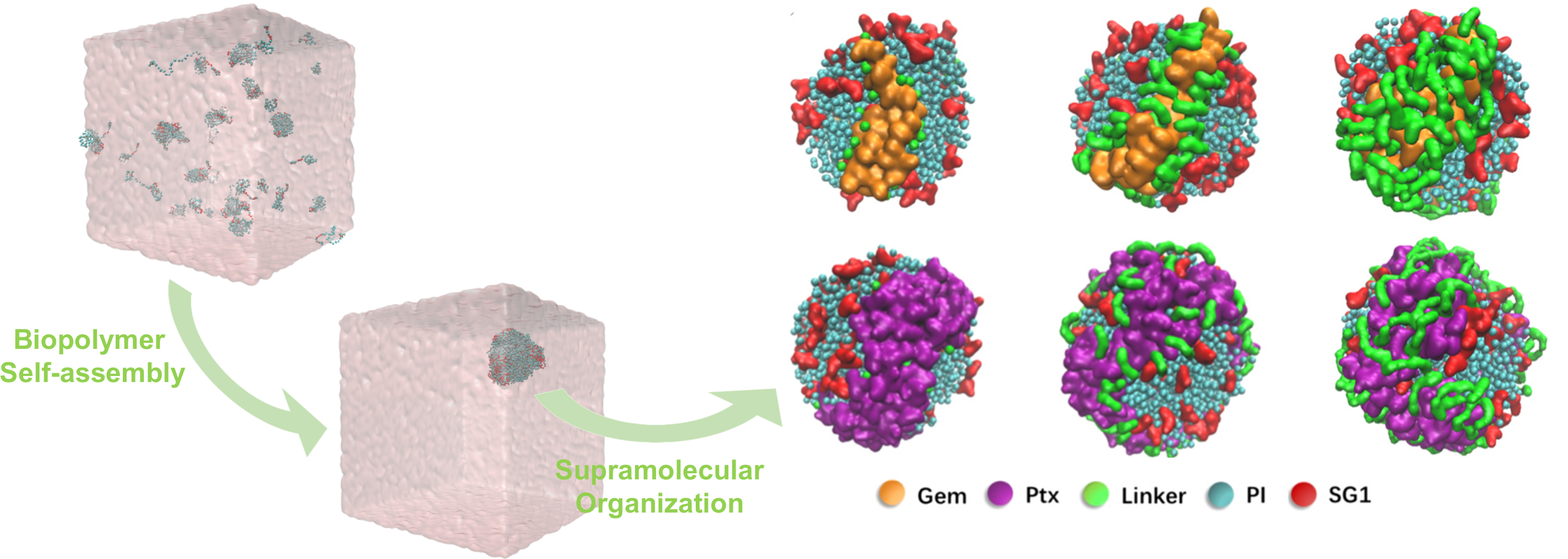
En lien avec notre intérêt pour les protéines intrinsèquement désordonnées, notre groupe a établi de solides collaborations avec l’Institut Galien Paris-Saclay afin de contribuer au développement de biopolymères pour le chargement efficace de médicaments dans des nanocarriers et leur libération contrôlée dans des tissus ciblés. En utilisant des simulations avancées de dynamique moléculaire à gros grains, nous étudions le processus d’auto-assemblage et la séparation de phase liquide-liquide des biopolymères thermosensibles et nous examinons l’organisation supramoléculaire des nanoparticules de polymère médicamenteux. L’objectif de ces études récentes est de mieux comprendre les propriétés physico-chimiques et structurelles qui régissent la formation et la dégradation de ces nanoparticules de biopolymères.
Inhibiteurs Peptidomimétiques
Les interactions protéine-protéine (IPP) étant impliquées dans de nombreux processus physiopathologiques, la recherche d’inhibiteurs d’IPP est une stratégie prometteuse mais difficile dans la conception de médicaments. Au sein de l’équipe FLUOPEPIT, le groupe de modélisation moléculaire développe et applique des méthodes computationnelles pour concevoir et étudier des peptides et des peptidomimétiques qui (i) seraient susceptibles de se lier à une interface protéine-protéine ciblée avec une haute affinité et (ii) auraient une bonne stabilité protéolytique et une bonne perméabilité membranaire. Notre stratégie à cet effet est une approche basée sur les fragments qui identifie les séquences optimales de peptides cycliques pour la liaison d’une surface protéique spécifique. En outre, nous utilisons des simulations de dynamique moléculaire améliorées pour caractériser les ensembles conformationnels de divers peptidomimétiques (cycliques, fluorés, aza…) et leur perméabilité membranaire.


{kind=link}
{kind=link}