Accueil > Personnels > ALAMI Mouad
ALAMI Mouad
DIRECTOR of RESEARCH at CNRS & DEPUTY DIRECTOR BioCIS
Team Leader : Design & Synthesis of Bioactive Compounds
:contact contact me by email mouad.alami universite-paris-saclay.fr
![]() +33 1 46 83 58 87
+33 1 46 83 58 87
Mouâd ALAMI was born in 1959 in Fez (Morocco). He did his undergraduate studies at the University Pierre and Marie Curie in Paris and his PhD in 1987 at the same university with Pr. J.-F. Normant and G. Cahiez. After a two years (1987-89) post-doctoral position in collaboration with the Elf- Atochem company he joined on 1989 the CNRS (National Center for Scientific Research) at the Ecole Normale Superieure, ENS-Paris (Dr. G. Linstrumelle). In 1993, he spent one year of postdoctoral studies at the University of Santa-Barbara in the laboratory of Prof. B. H. Lipshutz and in 1997, he moved as an independent researcher at the CNRS to Cergy-Pontoise University. In 1999, he accepted a position at the, Faculty of Pharmacy (University Paris-Sud) where he became Director of research at the CNRS. He published more than 160 papers, 5 international patents and 10 book chapters. Dr. ALAMI, team leader within the UMR BioCIS, was a member of the Scientific Council of the Department of Chemistry and the National Committee of CNRS - Section 12 (2004-08) and in 2012 was appointed to the National Council of universities (CNU Section-32). Since 2008 he is a deputy director of the UMR 8076 – BioCIS
DR ALAMI’S FIELD OF SPECIALIZATION INCLUDE :
• THE DEVELOPMENT OF NOVEL SYNTHETIC METHODOLOGIES INVOLVING ORGANOMETALLIC CHEMISTRY : hydro and carbometallation reactions, selective C-C and C-N bonds formation in heterocyclic chemistry and polyunsaturated molecules, and the development of new multi-components reactions (MCR) towards organic synthesis,
• MEDICINAL CHEMISTRY particularly, the synthesis of structurally challenging and biologically relevant natural and non-natural products with antitumoral activity. We have a special interest in the design and synthesis of anti-mitotic agents that mediate their primary biological activity through a direct binding interaction with tubulin. Specific applications are in the discovery and development of vascular disrupting agents (VDA) for the treatment of solid tumors and ophthalmologic disorders. Additional projects include anti-angiogenic agents, novel hsp90 inhibitors, particularly those targeting C-terminal domain, as well as progesterone receptor antagonists.
SOME OF THE SYNTHETIC METHODOLOGIES IN WHICH WE CURRENTLY HAVE ACTIVE PROGRAMS INCLUDE :

An unprecedented directing effect (ortho-directing effect (ODE) or syn-direct offect (SDE)) in conjugated alkynes (diarylalkynes, enynes, enediynes) has been highlighted, and promotes regioselective addition of metal hydride (Sn, Si) to the triple bond, affording exclusively one isomer. The results are rationalized in terms of electronic polarization across the alkyne bond whatever the electronic nature of the ortho/syn substituent (electron-withdrawing or electron-donating)

Functionalization of alkynes under environmentally conditions is of great interest, because it allows a direct access to heterocyclic molecules, which are potentially useful for the discovery of new drugs.
METAL-CATALYZED SELECTIVE C-C BOND FORMIN PROCESSES
In recent years, our attention was paid to the discovery of novel methodologies that improve the efficiency, selectivity, and availability of starting materials and the environmental sustainability of synthetic processes.

We are particularly interested in using tosylhydrazones as coupling reagents in a new type organometallic-free metal-catalyzed C–C and C–N bond forming reactions. This transformation, which is still at an early stage compared to the state of the art in traditional cross-coupling methods, represents a very convenient methodology for the unconventional modification of carbonyl compounds.
Interest of this transformation was demonstrated with the synthesis of isocombretastatin series as promising vascular disrupting agents (VDAs), and 4-arylchromenes, thiochromenes of biological interest.
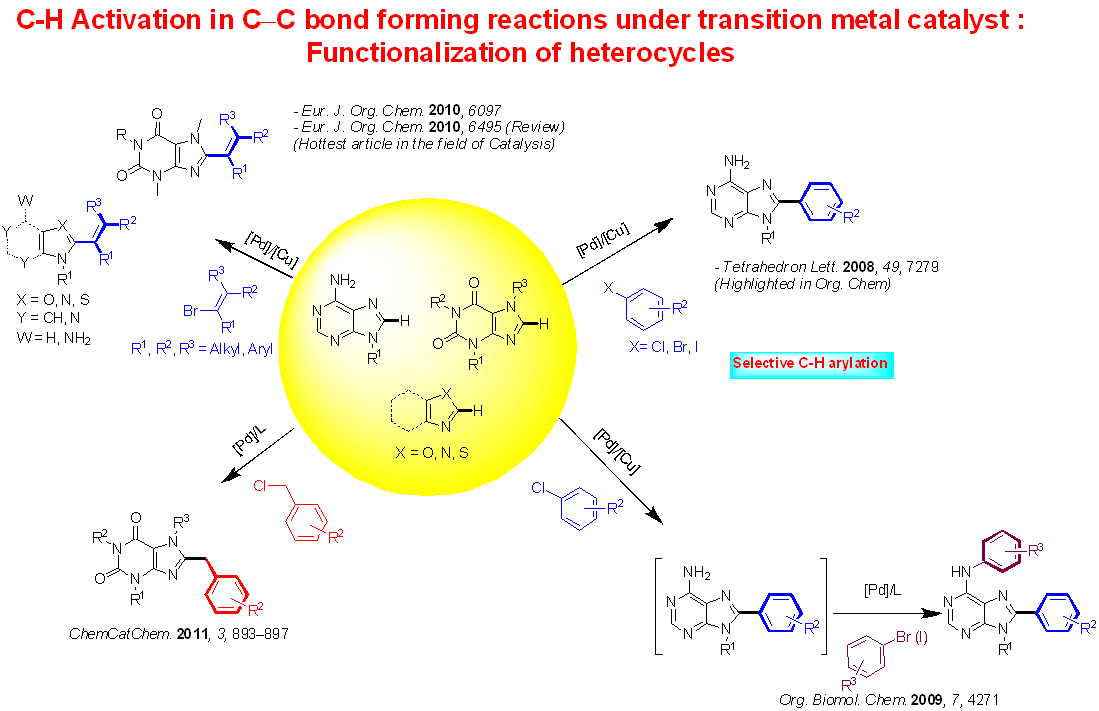
Within the field of heterocycles C-H functionalization, the majority of work has focused on a site selective C–H arylation of free-(NH2) adenines and application to the synthesis of 6,8-disubstituted adenines. The process based on the use of Pd(OH)2/C as the catalyst and a stoechiometric amount of CuI under ligandless conditions allows the coupling to proceed with a variety of electrophiles, including aryl, vinyl and benzyl halides (Cl, Br, I).

SOME OF THE MEDICINAL CHEMISTRY FIELD IN WHICH WE CURRENTLY HAVE ACTIVE PROGRAMS INCLUDE :

IsoCombretastatins A, having a 1,1-diarylethylene scaffold, constitute the simplest isomers of natural Z-combretastatins A, and are easy to synthesize without the need to control the olefin geometry. The discovery of isoCA-4 with biological activities comparable to that of CA-4 represent a major progress in this field demonstrating, that the 1,1-ethylene bridge is a nonclassical bioisostere for the Z-1,2-ethylene one. A number of these analogues showed activity in the low nanomolar range. A SAR of this series of compounds was carried out to provide a useful prediction for the design of further active ligands.

Heat shock protein 90 (Hsp90) is a significant target in the development
of rational cancer therapy, due to its role at the
crossroads of multiple signaling pathways associated with cell
proliferation and viability. A novel series of Hsp90 inhibitors
containing a quinolein-2-one scaffold was synthesized and
evaluated in cell proliferation assays. Results from these structure–
activity relationships studies enabled identification of the
simplified 3-aminoquinolein-2-one analogue 2b (6BrCaQ),
which manifests micromolar activity against a panel of cancer
cell lines. The molecular signature of Hsp90 inhibition was assessed
by depletion of standard known Hsp90 client proteins.
Finally, processing and activation of caspases 7, 8, and 9, and
the subsequent cleavage of PARP by 6BrCaQ, suggest stimulation
of apoptosis through both extrinsic and intrinsic pathways

Équipe
PUBLICATIONS
Lists of publications from 2000

CV - Pub - 1.3 Mio |